��������
��������
�������ƣ�
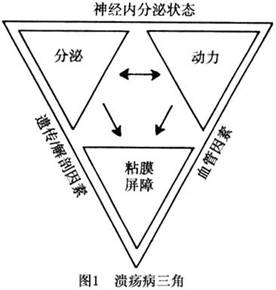
1.�������� SU��һ�ּ���θ��������������ķ������ƺ�SU�йأ����ֲ���ȫ��ͬ��SU���䱾���ķ����ص㡣Szabo(1984)������������ǣ��Ƚ������˵��θ�������������ļ�������֮��Ĺ�ϵ(ͼ1)��
(1)������ϵͳ�˷������ߣ�θ��Ӧ��״̬����Ϊ���е����٣�����������θ����ں�θ�䶯�����źͽ��ǿ�����θ�Ĥ���ã�����������������ʵ��֤���������������ľ���ϻ���������������ͬ�����������ڣ�Լ������Ȼ�����4��ı����ڣ����������½�����ˮ�У�2h��θ�Ĥ���������ü���Ѫ��ʵ�黹������������Ѫ��θ����ˮƽ���ߣ����ܺ��������˷��йء�������ϵͳֱ��Ӱ���θ����ں�θ������������;���´�嶯����������ǰ��-������ϵͳ�������Ժ�-������ϵͳ�������Ժ�-����-������ϵͳ����Ӧ��ʱ���Ƿ�ͨ��һ������һ�����ϵ�;�����в��������
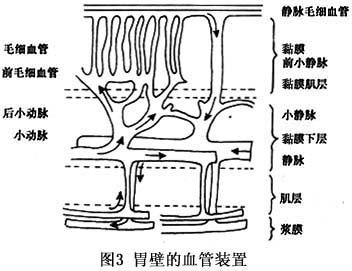
(2)θ�Ĥ���ϵ����ˣ���SU��˵����һ���dz���Ҫ�ķ���ԭ��Ҳ����˵�Ƿ����ı�Ҫ������θ�Ĥ�ǹ���ʮ�ָ��ӵ���֯�ṹ�����Ĥ����鲼Ϊ���ڶ��θС����ÿ��С������������ϸ��ƿ״θ�ٵĿ��ڣ�θ�ٵ�����Ƥ�ɼ��ֲ�ͬ���ܵ�ϸ��������ɡ���ϸ������θ�ᣬ��ϸ������θ����øԭ������ƿ�����ı�״ϸ�������Һ����С����С��֮��Ϊ������Ƥϸ��������ϸ���ڸ���̼����ø���ܲ���HCO3������Ȼ�������ֻ��H����5%��10%���������������Ϊ300��400��mol/h������θ�Ĥͬʱ��������Ϳ���Ļ��������Լұ��������á�θ�Ĥ���ڵ�θ��-θ����ø�Ǹ�Ч�ܵ�����Һ����θ��ﵽ���Ũ��ʱ���Ĥ�����H���ݶȿ����1000�����������ѧ�İ�Ĥԭ����H����Ȼ������ɢ���Ĥ�ڣ�Ϊ�˳�ַ���HCO3�������ã��ͱ�������Һ�Ĺ��ܡ�θ�Ĥ���渲�ǵ��ҺΪŨ��30��50mg/ml���ǵ����壬���Ϊ0.5mm���䱾���������Է�ֹH��������������ʹ������Ƥϸ�����ڵ�HC03���������Һ�м��ۣ�������ʧ����������θǻ��ɢ����Ϊ��ֹH��������Ϯ����Ч�����(ͼ2)�����⣬��θ������ʱ������HCO3�������������⣬���ɳ��ּ(alkaline tide)����ά��ƽ�⡣
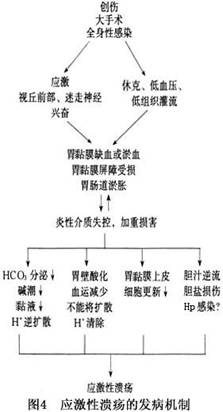
�Ĥ������Һ��θ��͵���ø���⣬���ֲ������µ��Һ���ڲ��䡣����θ������Ƥϸ���ڣ�θС������������Ƥϸ���������Ĥ������и��£���ÿ3���û�1�Ρ���Ƥϸ�������ֹ��ܻ�Ǹ����ܹ��̣���֤�书�ܵı�Ҫ������θ�ĤѪ����θ��Ѫѭ���ܷḻ����Ĥ��Ѫ�ܾ����㴩֧���Ĥ�²㣬�ڴ˲����������Ķ�������֧ͨ�������;����ֳ�С������С����������һ���ܱ����Ĥ����(muscularis mucosa)���ڴ˲���С������֧Ϊ��С��������֮��ǰëϸѪ�ܼ�ëϸѪ��ǰ��Լ��ȴ�����Ĥ���ΪëϸѪ�ܣ���ͬ��εľ�������(ͼ3)��
�κ�Ӱ��θ��Ѫ�������ض����θ�Ĥ��Ƥϸ���Ĺ��ܲ���Ӱ�죬����θ�Ĥ���ϡ������������ش��ˡ�ȫ���Ը�Ⱦ��Ӧ��״̬���ر����ݿ�����ĵ�Ѫ����ע�����ܼ���θ�ڵ�Ѫ��������SU��
(3)θ���H�������ã�θ���H��һֱ����Ϊ������������Ҫ���ء�θ��������Ȼ�ܼ���θ�Ĥ����ϵͳ�ĸ��ɣ���SUʱθ��һ�㲻�ߣ��������٣�������ˣ��Բ��ܷ�H����SU�����е����á�
����θ�Ĥ��������H��Ũ���䲻�ߣ��Կ�������ɢ������θ�����ữ������θ����pH����7.2���罵��6.5���£���ɲ�������θ�Ĥ��H���ɴ�ʹ��֯���ͷţ��̼������������θ��͵���ø�ķ������࣬��������ëϸѪ��ͨ�ԣ������Ĥ������Ƥϸ��HCO3���IJ�����ͬʱ��θ��Ѫ�����٣��ֲ��ܽ���ɢ��θ���ڵ�H����ʱ������������ؾ��ɵ���SU�ķ��������������ж�δ�ܾ�����Ҳ��ʹθ��pH�½�����SU�ķ�������Ӱ�졣
(4)��л�����Ӱ�죺θ�Ĥ����ϸ�����ر���θ�Ĥ������Ƥϸ����Ƶ�����£�ʹ��Ĥ��֬�Ĵ�л���������ϩ�����࣬��Ϊ�����ڻ�����ø�Ĵ��ºϳ�ǰ�����أ����в����϶��PGl
2��PGE
2�к�ǿ��������ԣ���θ�Ĥ�б������á�PGl
2������θ��ķ��ڣ�����θ�ĤѪ�����������ĤHCO
3�����Һ�ķ��ڣ���ֹH��������ɢ��PGE
2����Ѫ�ܣ��Ը���θ�ĤѪѭ����������Ϊ���ԡ���θ�Ĥ�У�PGl
2�ĺ���Ҫ��PGE
2��10�����ϡ�����ǰ�����صı������ã���֮ΪPG-�鵼����ϵͳ����˾ƥ�ֺ;ƾ�֮�������շ�
����θ�Ĥ�����������������ǿ�������ֹPG�ĺϳɡ�����ʵ���ȸ�����ιʳPGE
2���ٸ��谢˾ƥ�֣����Է�ֹSU�ķ�������Ӧ��״̬�£��ر���θ�ĤȱѪ��ȱ��������£�PG�������٣����һ����������һЩ���Խ��ʵ�ʧ�أ������ڻ�����ø���ü���PG�������ٵ�ͬʱ������5-֬����ø���õļ�ǿ��������ϩ��ת����лΪ����ϩ��ѪС�弤������(PAF)������ϩ��һ������Ѫ�����ʣ�PAF�оۼ�ѪС������ã���Щ���ʵIJ�����������θ�ĤȱѪ��ȱѪ��������θ�Ĥ�µķʴ�ϸ��(MC)Ҳ�ɲ���һЩ���ʣ�MC�����ɽ����֯�кܳ�����ϸ��������СѪ�ֲܷ�����һ�ֶ����̼�ʮ�����е���֯��Ӧϸ��������������翹ԭ�Ӵ��������IJ�λ����Ƥ��������������������MC�������ر�θ�Ĥ����Ϊ���ڶ��MC���ܵ�Ӧ����ȱѪ�ȴ̼��ɲ��������������ͷ���֯������ϸ���������ӡ����ء�����ϩ��PAF�ȣ����кܶ�������ʾ��ɴ�ʹSU�ķ�����������֯��������ëϸѪ��ǰ��Լ����С�������������Ĥ��ëϸѪ�ܣ���ʹθ�Ĥ�³�Ѫ����ʹ�ĤȱѪ����֯�����ɴ̼�θ����ڣ������Ĥ��Ѫ��ͨ�ԡ�������������Ƥϸ��DNA���ø�Ļ��ԣ�������Ƥϸ���ĸ��¡�����ʵ��Ҳ֤����̼������ɳ��ַʴ�ϸ���ġ����������ĤȱѪ�ĺ����ëϸѪ����Ƥϸ��������������л�����NO(��Ƥϸ�������ɳ�����EDRF)��ǿ�ҵ�Ѫ���������ʣ�����ѭ����ȫͣ�ͣ�ʹ�Ĥ��Ϊ�������ԡ�
(5)�����ݸ˾� (Hp)��Ⱦ����1983������Ի��θ�ײ��˵�θ�Ĥ��������Hp�Ժ���������θ�Ĺ�ϵһֱ�������ǵ���Ȥ��Hp�ɲ���ǿ���Ե���ø��ʹθ�Ĥ��Ƥ��������ؿ��ٷֽ⣬����NH3����NH3Ϊ�����ԣ���Hp�ܲ���ʹճ���Ǿۺ���ֽ��ø���ƻ������Ĥ������Һ�㡣����ܲ�����������ø����֬ø������ˮ��ø�Ⱦ���������Ƥϸ�����ݱ���ʮ��ָ��������Hp�ļ����Ϊ85%��θ������Ϊ53%����Hp��������IJ�ԭ��������������IJ����������ȷ��������ΪHp����ɼ���θ�ף������ٲ�����������Ҳ�����������Գ�Ѫ��
(6)���ε����ã����ٸ���������������Ĥ����ɥʧ�������������ԣ���θ��ȫ�г�������ؽ��ɵ�����Ķ�·�������ǹ����ԣ���θ�������ţ�Խ������ָ���˳���IJ��ˣ���������Һ����������Խ�������������������Ժ�����ԭ������ڡ����ζ�θ�Ĥ�����ò��ݺ��ӣ����α���Ϊ�dz���˾ƥ�ֺ;ƾ��������θ�Ĥ�����е�3λ�����ʡ����οɽ���θ�Ĥ�����е�3λ�����ʡ����οɽ���θ�Ĥ�ĵ�λ�����θ�Ĥ��ͨ�ԣ�ʹ��������H����Na�������״��Ĥ����ɢ�����ζ��Ĥ��Ƥϸ��Ĥ��֬�����ܽ����ã�������θ�Ĥ������ʵ��֤��ţ�ǵ������������θ�Ĥ�ķ��ڣ����������Ĥ��Ƥϸ����ATPø��
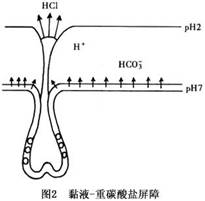
�Ӳ���Ƕ����������SU�����������ۺ����صĽ��(ͼ4)����ijЩΣ�ػ�����˵������������ΪSUʵ������
�����ٹ����ϰ��ۺ���(MODS)��θ(��������)�ı��֡�
2.�������� ����θ�Ĥ����ȷ�Ϊ���ú���������ָ��dz�IJ��䣬����Ȳ�Խ����Ƥ��Ļ���Ĥ��������Ļ�����Խ������Ĥ���Ĥ�£����뼡�㣬�������������Խ�����㣬��������������ּ���Ĥ�¼���Ĥ(ͼ5)��
������dz�㣬�����г�Ѫ�����������Ĥ�¼����㣬���ַ���������¶��Ѫ�������ɴ��Ѫ��
���Ĥ���������⣬�����ڽ���֯����֢�̶�Ҳ���в�ͬ������θ�Ĥ������ϸ�����������ڣ���������ϸ�������ܰ�ϸ������ϸ��������ϸ���������٣�ÿ�߱���Ұ��2��5�����ڱ�����Ƥϸ��֮��ż�ɼ��ܰ�ϸ��������Ϥ����࣬����θ�Ĥ��������Ƥ���ѣ������������������︲�ǡ���Ƥ����������ϸ���������������д�������ϸ������������ϸ��ռ�������е�״��Ѫ��Ҳ���е�״ŧ�Բ���(pit abscess)������������ص��Ǵ�����������ϸ������������ϸ���������ӣ����в�ͬ�̶���ά����
���ڲ������Ŀ��λ�ü���һ�¡����ò���㷺�ֲ���θ�Ĥ������ɶ��Ҳ�ɺ����ò��Ӳ��档����һ����θ���أ�θ�岿��֮������ȫ��θ�Ĥ���в��䡣���rͥ����Ϊ��������˺ϲ����Դ��Ѫ��������ʱ����θ�弰θ�Ĥ��Ƭȱ��ֱ����10cm���Ĥ����¶���������س�Ѫ������ɲ���ʳ���¶˺�ʮ��ָ���������ᷢ������ʮ��ָ����ʳ�ܲ����θ�Ĥ��������ߡ�
����
���ƣ������Ǵ���ԭ�����������ά��θ��pH��4.0���ϡ��������´�ʩ��
1.����ȫ����� �ٲ�Һ����Ѫ���ָ���ά���㹻��Ѫ������
2.���Ƹ�Ⱦ��
3.������ö�θ�д̼���ҩ��簢˾ƥ�֡����ء�ά����C�ȡ�
5.�ֲ����� ����θ����������ϴ��θ����ע����ø������
��������
��Ѫø�ȡ����б�������ˮ���մ�ˮϴθ��ϴθ��θҺ������Ϊֹ��
6.�ھ���Ӧ�� θ����ֹѪ���ɲ��õ�������������ֹѪ�Լ�θ���µľֲ���ҩ�ȡ�
7.�������� ����ѡ���Զ���Ѫ����Ӱ��˨����ע��Ѫ������ҩ�����ѹ�صȡ�